Early diagnosis is then crucial for appropriate management,
avoidance of the aforementioned complications and surgical planning.
After a careful physical examination of the abnormal cranial shape,
plain radiography of the skull is used as a first imaging modality to confirm the suspected diagnosis.
Using anterior-posterior (AP) and lateral views,
prematurely fused sutures are easily identified by the:
- absence of sutures
- associated bony ridging of the suture line
- sclerotic margins
- Abnormal cranial and orbital shapes
- copper-beaten appearance of the calvarium
In addition to the findings above stated,
there can be situations which may not be apparent on plain films.
These include detecting partial suture fusion,
which is best assessed using tridimensional reconstructions,
and evaluation before three months old,
age in whichthereis inadequatemineralizationof thebone.
Moreover,
the accuracy in diagnosis is vital and computed tomography (CT) with tridimensional and maximum intensity projection (MIP) reconstructed images is pertinent in inconclusive cases,
presurgical planning and intracranial associated anomalies,
such as hydrocephalus,
cerebral malformations or venous drainage anomalies.
Particularly,
the reformations allow evidence that may not be readily appreciated on axial CT images,
improving diagnostic accuracy and interobserver agreement.
However CT scans involve ionizing radiation exposures,
a matter of particular concern for pediatric populations because of their longer remaining life expectancy during which cancer may potentially develop and intrinsically greater tissue radiosensitivity in comparison with adults.
Postnatal ultrasonography is of limited value in the actual assessment of craniosynostosis but several studies have shownsome promising results in the evaluation of the lambdoid suture.
Magnetic Resonance (MR) imaging is performed only when malformations are suspected clinically or on CT,
particularly in syndromic or multisutural synostosis,
enabling superb intracranial soft tissue detail.
Types of Synostosis
Different names are given to the various types of craniosynostosis,
depending on which suture or sutures are involved,
including the following:
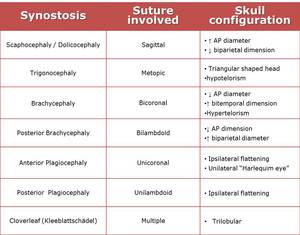
Table 2: The relationship between the various locations of craniosynostosis and the resulting skull configurations.
Scaphocephaly
Premature fusion of the sagittal suture is the most common isolated craniosynostosis,
constituting approximately half of all cases.
As a result of the fusion,
the infant's head does not expand in width but grows long (dolichocephaly) in the anterior-posterior dimension and narrow in the bitemporal direction,
producing a boat-shaped head (scaphocephaly) with frontal bossing and a palpable ridge along the closed suture [6] - figs.
2-5.
In spite of a tendency for speech disorders,
neurologic deficits and increased ICP are rare.
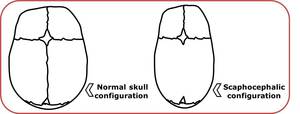
Fig. 2: Comparison between normal and scaphocephalic configuration.
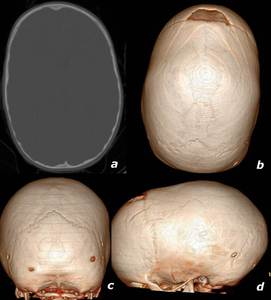
Fig. 3: 25 month old, male, with sagittal synostosis. a: axial CT in bone window. c-d: tridimensional volumetric reconstructions obtained using a 64-slice multidetector CT scanner demonstrate an elongated configuration of the skull with increased AP dimension. Note that lambdoid (c) and coronal (d) sutures have normal interdigitations.
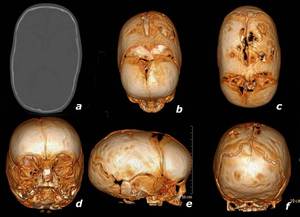
Fig. 4: 2 month old, male, with partial sagittal synostosis. a: axial CT in bone window. b-f: tridimensional volumetric reconstructions (64-slice multidetector CT). These images show partial premature fusion of the sagittal suture in its anterior segment («), finding not noticeable on plain films. Remaining sutures are patent.
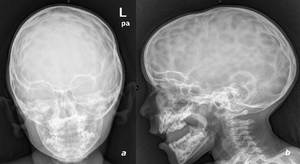
Fig. 5: 4 years and 9 month old, female. Frontal (a) and lateral (b) radiographs reveal a copper beaten skull appearance, associated with raised intracranial pressure, in a child with increased AP diameter assuming a scaphocephalic configuration (b).
Trigonocephaly
Trigonocephaly results from premature ossification of the metopic suture before approximately 6 months of age (figs.
6-9).
The characteristic imaging feature of metopic craniosynostosis is a quite pointed forehead,
like a triangle,
with flattened frontal bones and compensatory bossing of the parieto-occipital regions.
Typical premature metopic suture fusion occurs from the glabella towards the bregma,
producing incomplete forehead development and closely placed eyes (hypotelorism).
Other features that can be perceived are hypoplasia of the ethmoid sinuses with protrusion of the medial and retraction of the lateral orbital rim [6,7].
In recent years,
milder forms of this condition when the suture fuses after 6 months,
are termed “metopic ridge” and usually don`t imply surgical treatment.
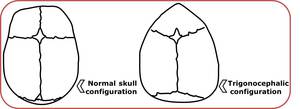
Fig. 6: Comparison between normal and trigonocephalic configuration.
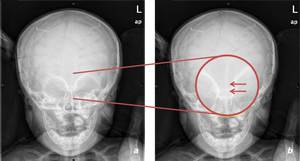
Fig. 7: 7 month old, male, with trigonocephaly. a, b: AP radiograph with an amplified area revealing metopic suture marginal sclerosis and hypotelorism.
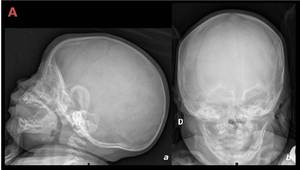
Fig. 8: 2 month old, male. Lateral (a) and frontal (b) plain films illustrate minor anterior triangular shaped head raising the suspicious for trigonocephaly.
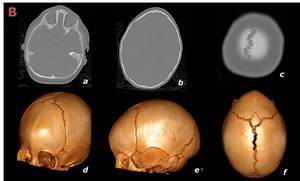
Fig. 9: Same infant as in figure 8. a-c: axial CT in bone window. d-f: tridimensional volumetric reconstructions (64-slice multidetector CT). The diagnosis was confirmed by metopic suture fusion with ridging (d,f).
Brachycephaly
Premature fusion of both coronal sutures with a resultant increase in the biparietal dimension as well as restriction of anterior-posterior growth (figs.
10-12).
This deformity is often syndromic and since the coronal suture develops in conjunction with the sutures at the base of the skull,
mid and upper face hypoplasia can occur.
Harlequin appearance of the orbit,which is not an uncommon finding,
represents the elevation of the superolateral angle of the orbit along with a flat frontal bone and resembles the widely knownharlequin's mask [3,6,7].
Bilateral lambdoid synostosis also shortens the skull in the AP diameter,
producing posterior brachycephaly.
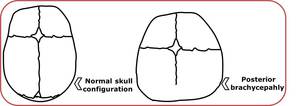
Fig. 10: Comparison between normal configuration and a skull with posterior brachicephaly.
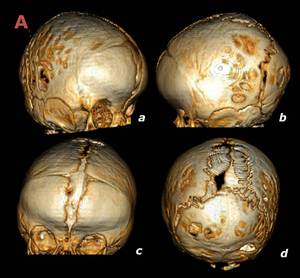
Fig. 11: 4 month old, female. Tridimensional volumetric reconstructions (a-d) exhibit partially fused coronal sutures with ridging.
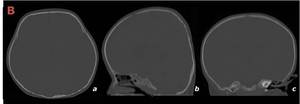
Fig. 12: Axial (a), sagittal (b) and coronal (c) CT in bone window of the same patient demonstrate a brachycephalic deformity with increased biparietal diameter (c) and reduced AP component (a). It also reveals a “tall” cranium (b) known as turricephaly.
Anterior plagiocephaly
Unicoronal synostosis (fig.13) resulting in anterior plagiocephaly presents with [3,6,7]:
- Flattening of the frontal bone on the affected side
- Contralateral frontal bossing
- Ipsilateral deviation of nasal root and nasal septum with contralateral deviation of nasal tip
- Ipsilateral harlequin eye deformity
- On the same side of fused suture ear lies anteriorly and superiorly
Fig. 13: Comparison between normal configuration and a skull with anterior plagiocephaly.
Posterior plagiocephaly
Posterior plagiocephaly can be triggered by early ossification of the lambdoid suturewhich is the least common ofthe primary synostosis.
As the concept 'plagiocephaly' refers to any flattening of the skull this condition must be distinguished from the more frequent positional molding that is almost never treated surgically (figs.
14-15).
In deformational or positional plagiocephaly no synostosis is identified and since the American Academy of Pediatrics in early 1990`s recommended that infants should sleep on their backs or sides to reduce the risk of sudden infant death syndrome,
has been seen with dramatic increased frequency. It is then caused by compressional forces such as intrauterine pressure/restrictive uterine environment,
postnatal preferential head positioning in infants sleeping,
overuse of infant seats.
It is then caused by compressional forces such as intrauterine pressure/restrictive uterine environment,
postnatal preferential head positioning in infants sleeping,
overuse of infant seats.
In positional plagiocephaly,
in addition to a non-fused lambdoid suture,
CT scan features include among other,
frontal bulging ipsilateral to the parieto-occipital flattening.
Viewed from above,
the head shape in positional molding resembles a parallelogram,
whereas that in lambdoid craniosynostosis is trapezoid shaped (fig.
16).
The key differences betweenunilateral lambdoid synostosisand positional plagiocephaly are summarized in the chart [3,8,9].
Fig. 14: Comparison between normal configuration and a skull with posterior plagiocephaly.
Fig. 15: Comparison between normal configuration and a skull with positional or deformational plagiocephaly.
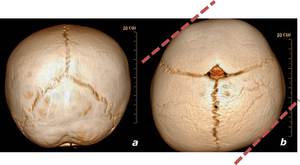
Fig. 16: 3D reconstructions (a,b) from a male with 4 months old. Left posterior plagiocephaly with no recognisable synostosis. The skull shape resembles a parallelogram (b).
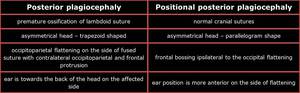
Table 3: Analysis of synostotic plagiocephaly versus positional plagiocephaly findings.
Cloverleaf skull (Kleeblattschädel)
Cloverleaf skull,
also known as Kleeblattschädel,
is an anomaly characterized by a trilobular skull with synostosis that may involve the coronal,
lambdoid,
and metopic sutures with bulging through an open sagittal suture or squamosal suture (fig.17).
Patients usually have serious neurological impairment.
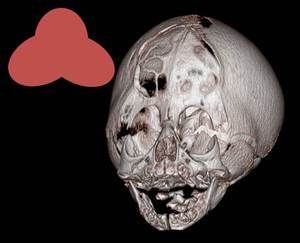
Fig. 17: Female, 6 days old. 3D reconstruction with a schematic drawing of a vague cloverleaf deformity with bulging of the temporal regions.
Syndromic Craniosynostosis
There are at least 180 known syndromes associated with synostosis,usually with autosomal dominant transmission and accompanyingother midline or skeletal abnormalities such as midfacial hypoplasia and limb abnormalities.
Crouzon and Pfeiffer syndromes are among the most frequentand share overlapping features.
Crouzon Syndrome (figs.
18-20)
This is the most common of the syndromes representing approximately 5% of all craniosynostosisand is thought to be due to a genetic mutation inFGFR2,
set on the short arm of chromosome 10 (10q26).
It is commonly linked with bilateral coronal craniosynostosis,
midfacial abnormalities as hypoplastic maxilla,
hypertelorism and exophthalmos.
There generally are no limb abnormalities associated.
About 30% of patients will have hydrocephalus with and without chronic tonsillar herniation,
but intelligence is often normal [3,6].
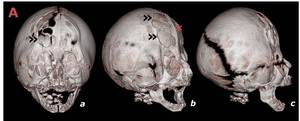
Fig. 18: a-c: Tridimensional reconstructions obtained using a 64-slice multidetector CT from a 6 days female with known Crouzon`s syndrome. A midline bony ridge (*) is evident at the metopic level with partial coronal synostosis. Areas of calvarial thinning (Luckenshaedel - ») with bony gaps are also visible.
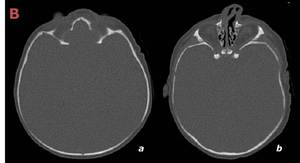
Fig. 19: Same infant as in figure 17 with axial CT in bone window (a,b). It is showed the increased biparietal diameter.
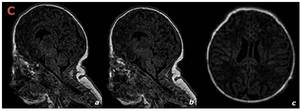
Fig. 20: Same infant as in figures 17 and 18. The sagittal (a,b) and axial (c) MR images – 1,5T – demonstrate normal brain parenchyma as well as no associated tonsillar herniation.
Pfeiffer Syndrome (figs.
21-22)
This syndrome is also characterized by craniosynostosis but now with accompanying limb deformities.
Similarly to Crouzon,
hydrocephalus is common as well as midfacial abnormalities and bicoronal synostosis.
It is inpart due tomutations in the FGFR1 or in the FGFR2 gene [3,10].
Several types withdifferent clinicalcourse andprognostic implicationshave been described:
- Type 1,
the classic form and least severe form,
is life compatible and consists of craniosynostosis,
hearing loss,
midface deficiency,
broad thumbs,
great toes and syndactyly.
- Type 2 involves a cloverleaf skull conformation with severe proptosis and hands-feet extension.
- Type 3 does not comprise cloverleaf skull.
There is often mental retardation and hydrocephalus.
In type 2 and 3 outcome is unfavorable with poor prognosis.
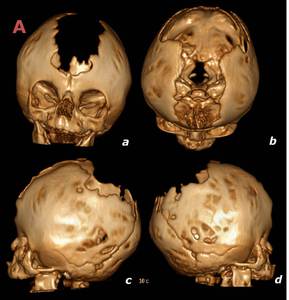
Fig. 21: 3 month old, female. Tridimensional volumetric reconstructions (a-d) in a patient with Pfeiffer syndrome show ossification of the coronal sutures accompanied by a wide midline defect.
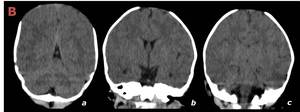
Fig. 22: Same patient exposed in figure 20 but with brain parenchyma window (a,b). There are no identifiable intra-parenchymal alterations.
